第一性原理计算已被广泛应用于物理、材料、化学、生物相关的科学研究。然而,受限于计算效率和精度,如何实现大尺度材料体系的第一性原理研究是该领域的一个重大挑战。基于人工神经网络的深度学习方法为解决该挑战问题带来了曙光。近期,深度学习已经成功应用于精确预测原子间相互作用,并加速分子动力学模拟。相比之下,理解、预测材料物性离不开电子结构计算,其深度学习的方法实现更具挑战性,研究进展有限。因此,发展深度学习方法、解决第一性原理电子结构计算的效率-精度两难困境是一个关键的科学问题。
betway必威徐勇、段文晖研究组发展了一种深度学习方法DeepH,能实现由材料构型快速、高精度预测任意原子构型的密度泛函理论哈密顿量,可极大加速电子结构计算。DeepH方法充分利用了电子性质的局域性原理,只需要利用小体系数据集训练的模型便可以在大尺度材料体系给出准确预测。通过引入局域坐标系与基组变换,DeepH方法能妥善处理哈密顿量的旋转协变问题,确保模型的泛化能力并简化学习难度。DeepH方法从密度泛函理论数据中学习,并预测需要研究材料构型的哈密顿量,可跳过耗时的密度泛函理论自洽迭代过程,实现高效的第一性原理电子结构计算。
通过对多种代表性材料的态密度、能带结构、非线性光学响应多种物理性质的精确预测,该工作展示了DeepH方法的高精度(预测误差为毫电子伏特级别)和良好的可迁移性。测试材料体系涵盖了准一维/准二维/三维的空间维度、平坦或卷起的原子构型、强化学键或范德瓦耳斯键、单种或多种化学元素、考虑了有或无自旋轨道耦合的情况,充分说明了DeepH方法的通用性与可迁移性。DeepH方法的一个成功应用是研究双层转角范德瓦尔斯材料。转角材料具有丰富的新奇物理性质,但因为该体系具有很大的摩尔超周期,传统的密度泛函理论计算难以研究其电子结构。DeepH方法只需通过无转角材料构型的密度泛函理论结果训练,就可以高效准确地预测各种转角材料构型的物理性质,这一流程不受限于元素种类和单胞形状,可用于构建转角材料高通量数据库。DeepH方法可以为大尺度材料和物理问题的第一性原理研究提供新契机。
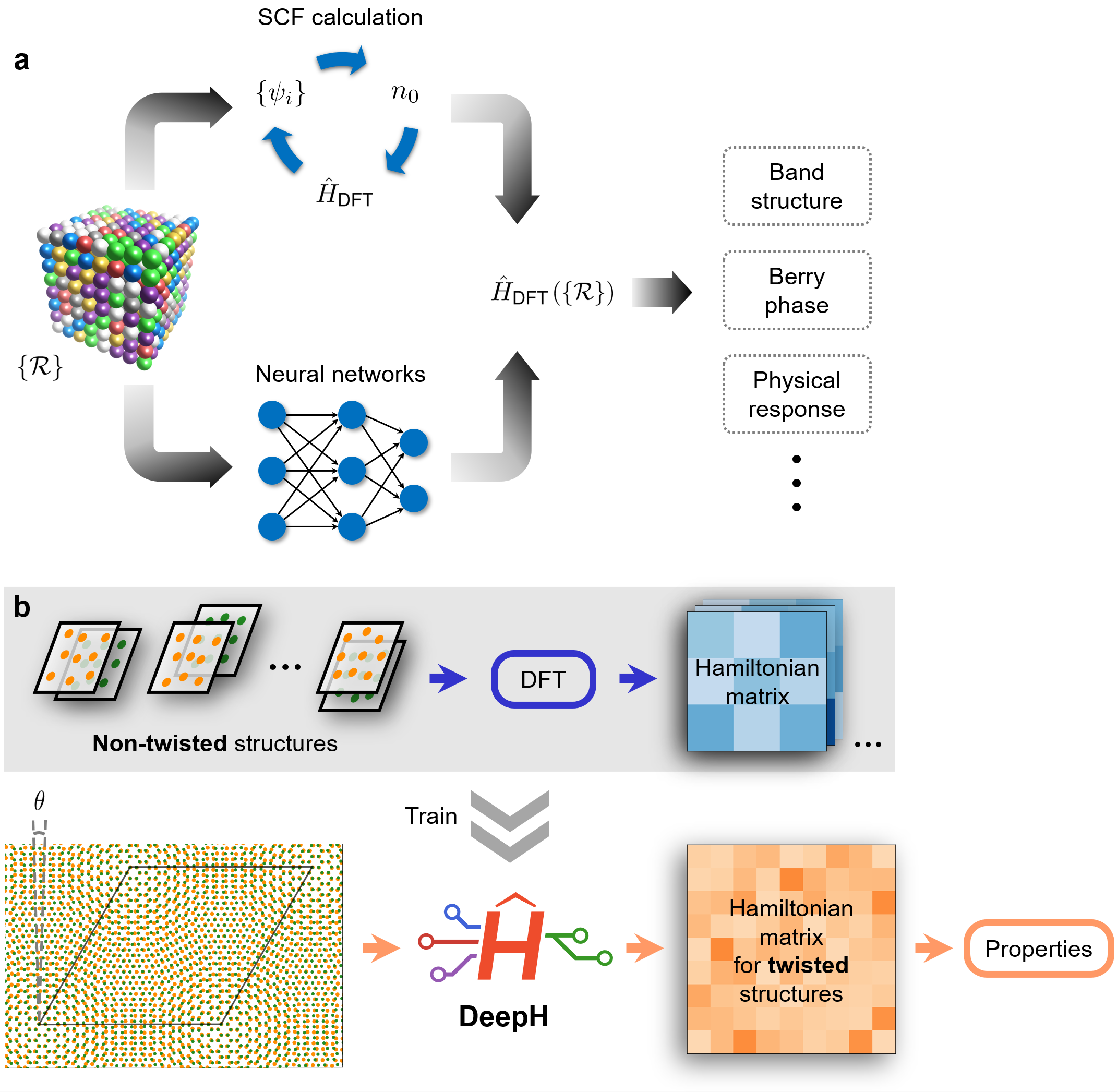
DeepH方法用于高效电子结构计算的示意图。a, 密度泛函理论哈密顿量是材料构型的函数,通常由耗时的自洽计算得到,DeepH方法利用深度神经网络预测给定原子构型的哈密顿量,得到的哈密顿量可以被用于计算各种物理性质。b, 使用DeepH方法研究转角范德瓦尔斯材料的流程图。通过无转角材料构型的密度泛函理论结果训练,DeepH模型可以高效准确地预测各种转角材料构型的物理性质。
该成果以“Deep-learning density functional theory Hamiltonian for efficient ab initio electronic-structure calculation”为题发表在6月23日的《自然·计算科学》(Nature Computational Science)。同期,该杂志还发表了以“Improving the efficiency of ab initio electronic-structure calculations by deep learning”为题的Research Briefing介绍上述成果。
betway必威徐勇教授和段文晖教授为该论文的通讯作者,研究组博士生李贺、王尊、邹念龙为共同第一作者。合作者还包括研究组博士后叶萌、徐润章和北京大学本科生贡晓荀。该工作得到了国家自然科学基金委基础科学研究中心、国家杰出青年科学基金、国家科技部等项目单位的支持。
文章链接:https://www.nature.com/articles/s43588-022-00265-6
Research Briefing链接:https://www.nature.com/articles/s43588-022-00270-9
该工作开发的DeepH程序包已作为开源软件供用户使用,用户可在下方链接获取开源仓库和使用手册。
https://github.com/mzjb/DeepH-pack https://deeph-pack.readthedocs.io

